
Glikacja jest nieenzymatyczną reakcją powstawania wiązań kowalencyjnych między cząsteczkami cukrów - takich jak glukoza lub fruktoza - a cząsteczkami białek i lipidów. Glikacja białek, dotyczy łańcuchów bocznych lizyny, a jej skutkiem są często patologiczne modyfikacje białek, powodujące utratę części bądź całości ich aktywności biologicznej. Najlepiej poznaną i najobszerniej opisaną klasą receptorów końcowych produktów zaawansowanej glikacji są receptory RAGE, których rola w wielu stanach patologicznych organizmu jest coraz bardziej podkreślana.
Na wstępie warto zaznaczyć, że glikacja jest procesem odmiennym od enzymatycznych reakcji glikozylacji, które stanowią jeden z kluczowych elementów potranslacyjnej modyfikacji białek mających na celu powstanie ostatecznej biologicznie aktywnej formy tych cząsteczek. W procesie glikacji zaś, białka podlegają złożonym przemianom zwanymi reakcją Maillarda. W końcowych etapach reakcji Maillarda, w wyniku kondensacji i sieciowania, powstają w sposób nieodwracalny końcowe produkty zaawansowanej glikacji (AGE – advanced glycation end products). Produkty te tworzą bardzo heterogenną grupę związków o zróżnicowanych masach cząsteczkowych. Pomimo, że powstawanie takich związków zdefiniowano i opisano już ponad 90 lat temu, dokładna budowa większości z nich wciąż jest słabo poznana. Poziom AGE w organizmie zależny jest nie tylko od szybkości ich wytwarzania, ale również od tempa, z jakim usuwane są z ustroju. W warunkach homeostazy podlegają one rozkładowi w lizosomach komórek, a produkty degradacji są wydzielane do krążenia i wydalane z moczem [1]. Pewna ilość AGE może być także usuwana z krążenia przez wątrobę i śródbłonek tej tkanki [2].
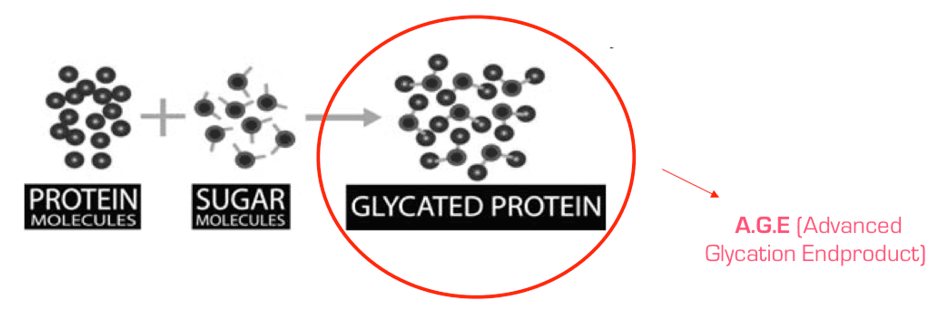
Glikacja w warunkach równowagi i w stanach jej zaburzenia
Proces glikacji może zachodzić zarówno wewnątrz komórek jak i pozakomórkowo, we wszystkich tkankach i płynach ustrojowych. Należy podkreślić, że zjawisko to dotyczy nie tylko białek, ale również DNA i lipidów [3]. Powstające w warunkach fizjologicznych AGE mają znaczenie regulacyjne. W życiu płodowym obserwuje się duże ilości glikowanych białek w komórkach macierzystych. Wykazano, że podczas różnicowania komórek są one szybko eliminowane w proteasomach (strukturách, w których dochodzi do degradacji białek). Badania sugerują, że wysoki poziom tak zmodyfikowanych struktur pełni ważną rolę w utrzymywaniu stanu komórek niezróżnicowanych [4]. W warunkach fizjologicznych proces glikacji jest procesem powolnym, przebiega przez całe życie i stopniowo prowadzi do starzenia organizmu [5]. Modyfikacji ulegają głównie białka o długim okresie półtrwania, takie jak kolagen, krystalina soczewki i albumina osoczowa [6]. Wiązania krzyżowe obecne w AGE prowadzą do wzrostu sztywności białek.
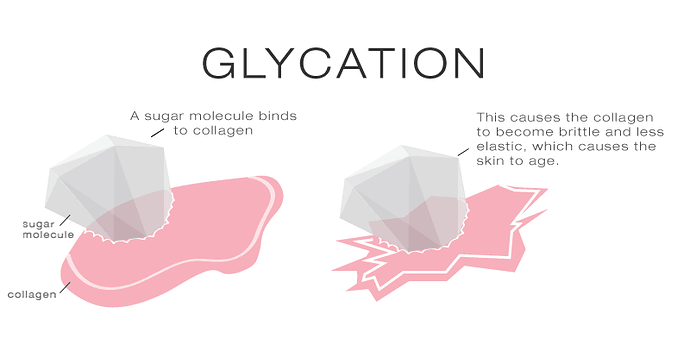
Z drugiej zaś strony, nasilony proces glikacji prowadzi do powstania nadmiaru wysokospolimeryzowanych i usieciowanych białek. Tracą one wówczas swoje biologiczne funkcje, stają się częściowo oporne na degradację proteolityczną, co znacznie utrudnia ich usuwanie z komórek i tkanek. Kumulacja takich produktów w postaci złogów powoduje także usztywnienie ścian naczyń oraz tkanek [7]. Badania naukowe wykazały postępujący wraz z wiekiem wzrost glikacji białek tkanki łącznej w ścięgnach, aorcie oraz soczewce oka. Zjawisko to ma zatem bezpośredni udział w rozwoju chorób związanych z wiekiem [1]. Dodatkowo, modyfikacja ta osłabia także strukturę jednego z najważniejszych białek w organizmie – kolagenu. W wyniku glikacji dochodzi do pogrubienia fibryli wchodzących w skład włókien i wzrostu liczby wiązań krzyżowych pomiędzy jego cząsteczkami, co skutkuje zwiększoną sztywnością, kruchością, obniżoną rozpuszczalnością i wrażliwością na procesy naprawcze. W związku z tym usuwanie powstałych wiązań jest utrudnione, a procesy naprawcze nieefektywne. Według przeprowadzonych badań skóra diabetyków starzeje się nawet o 30% szybciej niż skóra osoby zdrowej.
Zmiany patofizjologiczne stymuluje dodatkowo stres oksydacyjny towarzyszący powstawaniu AGE. Oczywistym jest fakt, że glikacja nasila się w stanie ostrej i chronicznej glikemii [9]. Dlatego w cukrzycy znacznie wcześniej dochodzi do akumulacji AGE w krążeniu oraz w różnych tkankach i narządach. Stwierdzono odkładanie się takich produktów w skórze, płucach, nerkach, jelitach, dyskach rdzenia kręgowego, w sercu i naczyniach nie tylko u osób w starszym wieku, ale także u pacjentów cukrzycowych [10]. Obserwowany w stanie hiperglikemii wzrost poziomu produktów zaawansowanej glikacji w różnych tkankach wyraźnie korelował z rozwojem późniejszych powikłań. Najczęstszą przyczyną powikłań mikronaczyniowych we wczesnej cukrzycy jest pogrubianie błony podstawnej kapilar i hipertrofii macierzy zewnątrzkomórkowej. Stanowi to podłoże rozwoju retinopatii, nefropatii, neuropatii cukrzycowej [11], natomiast komplikacje makronaczyniowe są przyczyną choroby arterii wieńcowych, choroby naczyń obwodowych i naczyń mózgowych [12].
Glikowane białka a „pamięć metaboliczna” w cukrzycy
Licznie podejmowane próby kliniczne wyraźnie wykazały, że wczesna i rygorystyczna kontrola glikemii znacznie redukuje i opóźnia wystąpienie późnych powikłań związanych z makro i mikroangiopatią. Kluczową rolę w tym zjawisku odgrywa utrzymanie właściwego poziomu AGE-białek. Jak wykazały badania, przewlekły brak właściwej kontroli glikemii skutkuje powstaniem stosunkowo trwałych zmian w poziomie glikacji białek strukturalnych i fukcjonalnych, a także zmiany epigenetyczne w jądrowym i mitochondrialnym DNA komórek chorych na cukrzycę [13]. Dodatkowo, prowadzone na zwierzętach cukrzycowych doświadczenia wykazały wyraźnie, że po kilkumiesięcznym okresie hiperglikemii, długotrwała normalizacja poziomu glukozy nie przywracała do normy wielu parametrów metabolicznych związanych z rozwojem powstałej angiopatii. Co istotne, pomimo powrotu do ścisłej kontroli glikemii poziomy potencjalnie szkodliwych związków takich jakAGE w mitochondriach, kolagenu w podścielisku tkankowym, kinazy białkowej C, NFκB, kaspazy 3, nadtlenków lipidów oraz rodników nitrozylowych pozostawały podwyższone, a poziom glutationu w dalszym ciągu pozostawał obniżony. Natomiast, skrócony okres niekontrolowanej hiperglikemii skutkował powrotem do normoglikemii, powodując częściowe odwrócenie powstałych zaburzeń metabolicznych [14]. Wyniki tych eksperymentów, wydają się wyraźnie tłumaczyć zależny od AGE-białek mechanizm powstawania powikłań naczyniowych w cukrzycy. Ponadto dane te dowodzą, że wiele zmian wywołanych glikacją ma charakter trwały i nie może być łatwo odwrócona przez późną kontrolę glikemii. Fakty te podkreślają zatem istotność jak najszybszego rozpoczynania terapii cukrzycy i prowadzenia jej w sposób rygorystyczny przez całe życie pacjenta.
Receptory RAGE
Powstawanie glikowanych agregatów białkowych niesie ze sobą dalsze konsekwencje, ponieważ AGE mogą się wiązać z receptorami na powierzchni komórek i wpływać na procesy wewnątrzkomórkowe [15]. Badania mechanizmów wychwytu AGE  przez komórki doprowadziły do odkrycia kilku typów receptorów powierzchniowych tych ligandów. Jednym z nich są receptory RAGE (ang. receptor for advanced glycation endproducts) będące wieloligandowymi receptorami błonowym. Połączenie liganda z receptorem RAGE skutkuje aktywacją wielu kluczowych szlaków sygnałowych wewnątrz komórki, które związane są z różnorodnymi stanami patologicznymi. Postuluje się udział receptora RAGE i białek z nim oddziałujących w postępie wielu schorzeń, m. in.: powikłania cukrzycowe (ligand: końcowe produkty glikacji), chorobie Alzheimera (ligand: peptyd Aβ), reumatoidalnym zapaleniu stawów (białko S100A12), przerzutów nowotworowych (białko S100P). Receptory RAGE zlokalizowano na powierzchni wielu typów komórek. Ich obecność potwierdzono m.in. na fagocytach, hepatocytach, komórkach śródbłonka i mięśni gładkich ściany naczyń, a także komórkach układu nerwowego [16]. W mięśniach szkieletowych szczurów wykazano, że tempo syntezy RAGE jest regulowane zależnie od etapu rozwoju organizmu, a jego nadekspresja powoduje przyspieszenie różnicowania mioblastów [17]. Zaobserwowano także, że biosynteza RAGE i jego liganda, amfoteryny, jest nasilona w neuronach rozwijającego się ośrodkowego układu nerwowego i zanika po urodzeniu [18]. W warunkach homeostazy ekspresja RAGE w komórkach innych tkanek, w tym w neuronach, fibroblastach, komórkach mięśni gładkich oraz w makrofagach jest niewielka. Ich indukcja nasila się jednak wskutek zwiększonej aktywacji komórek wywołanej wzrostem poziomu ligandów RAGE, w warunkach stresu, w stanach zapalnych, w cukrzycy, chorobie Alzheimera [19] oraz w reumatoidalnym zapaleniu stawów [20]. U osób chorych na cukrzycę, ze względu na wspomniany wcześniej zwiększony proces glikacji, dochodzi do zwiększenia syntezy RAGE w makrofagach i monocytach przenikających z układu naczyniowego do tkanek. Wykazano także, stymulowany przez zwiększony poziom AGE, wzrost ekspresji receptorów na śródbłonku i komórkach mięśni gładkich, skutkując ciągłym pobudzaniem komórek i nieodwracalnym uszkodzeniem tkanek [21].
przez komórki doprowadziły do odkrycia kilku typów receptorów powierzchniowych tych ligandów. Jednym z nich są receptory RAGE (ang. receptor for advanced glycation endproducts) będące wieloligandowymi receptorami błonowym. Połączenie liganda z receptorem RAGE skutkuje aktywacją wielu kluczowych szlaków sygnałowych wewnątrz komórki, które związane są z różnorodnymi stanami patologicznymi. Postuluje się udział receptora RAGE i białek z nim oddziałujących w postępie wielu schorzeń, m. in.: powikłania cukrzycowe (ligand: końcowe produkty glikacji), chorobie Alzheimera (ligand: peptyd Aβ), reumatoidalnym zapaleniu stawów (białko S100A12), przerzutów nowotworowych (białko S100P). Receptory RAGE zlokalizowano na powierzchni wielu typów komórek. Ich obecność potwierdzono m.in. na fagocytach, hepatocytach, komórkach śródbłonka i mięśni gładkich ściany naczyń, a także komórkach układu nerwowego [16]. W mięśniach szkieletowych szczurów wykazano, że tempo syntezy RAGE jest regulowane zależnie od etapu rozwoju organizmu, a jego nadekspresja powoduje przyspieszenie różnicowania mioblastów [17]. Zaobserwowano także, że biosynteza RAGE i jego liganda, amfoteryny, jest nasilona w neuronach rozwijającego się ośrodkowego układu nerwowego i zanika po urodzeniu [18]. W warunkach homeostazy ekspresja RAGE w komórkach innych tkanek, w tym w neuronach, fibroblastach, komórkach mięśni gładkich oraz w makrofagach jest niewielka. Ich indukcja nasila się jednak wskutek zwiększonej aktywacji komórek wywołanej wzrostem poziomu ligandów RAGE, w warunkach stresu, w stanach zapalnych, w cukrzycy, chorobie Alzheimera [19] oraz w reumatoidalnym zapaleniu stawów [20]. U osób chorych na cukrzycę, ze względu na wspomniany wcześniej zwiększony proces glikacji, dochodzi do zwiększenia syntezy RAGE w makrofagach i monocytach przenikających z układu naczyniowego do tkanek. Wykazano także, stymulowany przez zwiększony poziom AGE, wzrost ekspresji receptorów na śródbłonku i komórkach mięśni gładkich, skutkując ciągłym pobudzaniem komórek i nieodwracalnym uszkodzeniem tkanek [21].
Jest receptor – musi być i oddziałująca cząsteczka (ligand)
Receptory RAGE są wieloligandowe, wchodzą w interakcje i tworzą kompleksy z szeroką grupą zróżnicowanych strukturalnie białek. Bardzo istotnym jest fakt, że przyłączenie ligandów do receptorów RAGE aktywuje szlak kinaz MAP (kinazy aktywowane miogenami), które uczestniczą w wielu szlakach sygnałowych komórki. Aktywację szlaku kinazy ERK1/2(p44/p42) MAP wykazano w komórkach mięśni gładkich [22], miofibroblastach, mioblastach, osteoblastach [23] i monocytach [24]. Może to prawdopodobnie wynikać z bezpośredniej reakcji receptora RAGE z kinazą ERK [25]. Badania wykazały także wpływ RAGE na aktywację kinazy p38 i JNK/SAPK w komórkach nowotworowych i monocytach. Pobudzone oddziaływaniem RAGE szlaki sygnałowe skutkują aktywacją czynnika transkrypcyjnego NF-kB, który w cytoplazmie jest nieaktywny i związany z inhibitorem IkBa. Uwolniony aktywny czynnik NF-kB przemieszcza się do jądra komórki i aktywuje ekspresję genów cytokin (TNF-a, IL-1, IL-6) oraz białek adhezyjnych (VCAM-1, ICAM-1), które uczestniczą w procesach zapalnych [20]. Wykazano także, że w miejscach nadmiernego gromadzenia i akumulacji AGE w naczyniach dochodzi do wzmożonej ekspresji RAGE. Jest to mechanizm dodatniego sprzężenia zwrotnego - przedłuża działanie RAGE i nasila aktywację w ten sposób pobudzonych komórek. Zjawisko takie zaobserwowano m.in. w retinopatii cukrzycowej [28].
Nasilenie nagromadzenia się w organizmie AGE oraz RAGE zaobserwowane jest przede wszystkim w stanach patologicznych spowodowanych hiperglikemią. Ze względu na to, że glikacja nie jest reakcją wymagającą obecności enzymów, nie ma ścisłych ograniczeń i zachodzi ona tak długo, jak długo dostępny jest substrat. Liczne badania pozwalające zrozumieć zarówno proces glikacji, jak i oddziaływania ligandów (będących produktem tego procesu) z receptorami, umożliwiły wyjaśnienie pewnych szlaków sygnałowych prowadzących do pobudzania komórek. Poznanie tych mechanizmów ma ogromne znaczenie w definiowaniu skutecznych czynników blokujących interakcje ligand-RAGE, a co za tym idzie w zmniejszeniu skutków toksycznego działania AGE.
- Ahmed N., Thornalley P.J.: Advanced glycation endproducts: what is their relevance to diabetic complications? Diabetes Obes. Metab., 2007; 9: 233–245.
- Smedsrød B., Melkko J., Araki N., Sano H., Horiuchi S.: Advanced glycation end-products are eliminated by scavenger-receptor-mediated endocytosis of hepatic sinusoidal Kupffer and endothelial cells. Biochem. J., 1997; 322: 567–573
- Thornalley P.J.: Endogenous alpha-oxoaldehydes and formation of protein and nucleotide advanced glycation endproducts in tissue damage. Novartis Found Symp., 2007; 285: 229–243
- Hernebring M., Brolén G., Aguilaniu H., Semb H., Nyström T.:Elimination of damaged proteins during differentiation of embryonic stem cells. Proc. Natl. Acad. Sci. USA, 2006; 103: 7700–7705
- Ulrich P., Cerami A.: Protein glycation, diabetes, and aging. Recent Prog. Horm. Res., 2001; 56: 1–21
- Wa C., Cerny R.L., Clarke W.A., Hage D.S.: Characterization of glycation adducts on human serum albumin by matrix-assisted laser desorption/ionization time-of-fl ight mass spectrometry. Clin. Chim. Acta, 2007; 385: 48–60
- Sims T.J., Rasmussen L.M., Oxlund H., Bailey A.J.: The role of glycation cross-links in diabetic vascular stiffening. Diabetologia,1996; 39: 946–951
- Frye E.B., Degenhardt T.P., Thorpe S.R., Baynes J.W.: Role of the Maillard reaction in aging of tissue proteins. Advanced glycation end product-dependent increase in imidazolium cross-links in human lens proteins. J. Biol. Chem., 1998; 273: 18714–18719
- Bierhaus A., Schiekofer S., Schwaninger M., Andrassy M., HumpertP.M., Chen J., Hong M., Luther T., Henle T., Klöting I., Morcos M.,Hofmann M., Tritschler H., Weigle B., Kasper M., Smith M., Perry G., Schmidt A.M., Stern D.M., Häring H.U., Schleicher E., Nawroth P.P.: Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes, 2001; 50: 2792–2808
- Schleicher E.D., Wagner E., Nerlich A.G.: Increased accumulation of the glycoxidation product Ne-(carboxymethyl)lysine in human tissue in diabetes and aging. J. Clin. Invest., 1997; 99: 457–468
- Singh R., Barden A., Mori T., Beilin L.: Advanced glycation end products: a reviev. Diabetologia, 2001; 44: 129–146
- Rahman S., Rahman T., Ismail A.A., Rashid A.R.: Diabetes-associated macrovasculopathy: pathophysiology and pathogenesis. Diabetes Obes. Metab., 2007; 9: 767–78
- Li J.H., Wang W., Huang X.R., Oldfi eld M., Schmidt A.M., Cooper M.E., Lan H.Y.: Advanced glycation end products induce tubular epithelial-myofi broblast transition through the RAGE-ERK1/2 MAP kinase signaling pathway. Am. J. Pathol., 2004; 164: 1389–1397
- Andrzej Szutowicz. Hiperglikacja białek wewnątrz i zewnątrzkomórkowych; marker czy aktywny element patomechanizmów cukrzycy. Journal of Laboratory Diagnostics Diagn Lab 2015; 51(3): 213-220
- Schmidt A.M., Yan S.D., Yan S.F., Stern D.M.: The biology of the receptor for advanced glycation end products and its ligands. Biochim. Biophys. Acta, 2000; 1498: 99–111
- Brett J., Schmidt A.M., Yan S.D, Zou Y.S., Weidman E., Pinsky D., Nowygrod R., Neeper M., Przysiecki C., Shaw A., Migheli A., Stern D.: Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol., 1993; 143:1699–1712
- Sorci G., Riuzzi F., Arcuri C., Giambanco I., Donato R.: Amphoterin stimulates myogenesis and counteracts the antimyogenic factors basic fibroblast growth factor and S100B via RAGE binding. Mol. Cell. Biol., 2004; 24: 4880–4894
- Hori O., Brett J., Slattery T., Cao R., Zhang J., Chen J.X., Nagashima M., Lundh E.R., Vijay S., Nitecki D., Morser J., Stern D., Schmidt A.M.: The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of RAGE and amphoterin in the developing nervous system. J. Biol. Chem., 1995; 270: 25752–25761
- Lue L.F., Walker D.G., Brachova L., Beach T.G., Rogers J., SchmidtA.M., Stern D.M., Yan S.D.: Involvement of microglial receptor

Cynk a odporność – jakie są zależności?
Cynk największą popularność zyskuje jesienią. Jest to w pełni uzasadnione, ponieważ cynk pełni krytyczną funkcję w funkcjonowaniu układu odpornościowego. Jego…

Kurkumina a choroby nowotworowe – jakie są zależności?
Wizytówką kurkuminy jest jej działanie przeciwzapalne, które naukowcy bardzo szeroko opisują w publikacjach naukowych. Przewlekłe stany zapalne uznawane są za…

Magnez w sporcie – jakie są korzyści?
Im większa aktywność fizyczna, tym większe zapotrzebowanie na magnez. Jeśli chcesz zapewnić swojemu ciału optymalne warunki do uzyskiwania progresu sportowego,…

Koenzym Q10 a serce – jakie są zależności?
Serce nieustannie tłoczy krew, która zaopatruje wszystkie nasze tkanki w substancje odżywcze. Co będzie, gdy osłabi swoją pracę? Skutki są…

Maksymalna pompa mięśniowa
Właśnie rozpoczynasz przygodę z treningiem na siłowni, czy może jesteś doświadczonym zawodnikiem szukającym sposobów na optymalizację swojego treningu? Bez względu…

Posiłek potreningowy – najważniejszy w ciągu dnia?
W świecie fitness upowszechniło się takie przekonanie, które głosi, iż posiłek potreningowy jest najważniejszym posiłkiem jedzonym w ciągu całego dnia….

Strength & Conditioning – co to w ogóle jest?
Strength & Conditioning, czyli w wolnym tłumaczeniu siła i kondycjonowanie – co to w ogóle jest za dziedzina nauki i…

Długotrwały trening aerobowy a poziom testosteronu u mężczyzn
Jednym z fizjologicznych systemów organizmu, który jest niezwykle wrażliwy na stres związany z wykonywanymi systematycznie ćwiczeniami fizycznymi jest układ hormonalny….